Дмитрий Марфунин
"О кандидозе"
На главную
Как известно, Candida albicans (Candida) – это гриб-комменсал, обычно обнаруживается на поверхности слизистых оболочек влагалища, ротовой полости, в ЖКТ и на коже. «Комменсал» означает «сотрапезник», то есть организм, получающий пользу от другого организма, не причиняя ему вреда. Иначе говоря, он питается продуктами метаболизма организма хозяина. На коже такими продуктами могут быть липиды кожного сала, межклеточный матрикс и корнеоциты поверхностного рогового слоя эпидермиса. Действительно, Candida в состоянии комменсала конститутивно экспрессирует в небольшом количестве и фосфолипазу (PL) и липазу (LIP) [10, 15], которые расщепляют фосфолипиды и триглицериды, обеспечивая Candida свободными жирными кислотами. Однако для нормальной жизнедеятельности Candida необходим азот, источником которого служат белки. Но Candida в состоянии комменсала не экспрессирует протеолитические ферменты и, соответственно, не может расщеплять белки межклеточного матрикса и клеточной оболочки корнеоцитов. Кроме вышеназванных продуктов, в верхних слоях рогового слоя эпидермиса присутствуют гигроскопические свободные аминокислоты, обеспечивающие поддержание гидратации кожи и водную задержку, являясь главным источником естественного увлажняющего фактора (NMF) [20]. Можно думать о том, что именно свободные аминокислоты верхнего рогового слоя являются источником азота для Candida в состоянии комменсала.
Candida не прокариот, а эукариот, с большим геномом и, таким образом, большими ресурсами [11]. Способный выживать как сотрапезник в анатомически различных участках со своими собственными специфическими наборами средовых воздействий, Candida приспособил свой рост к диапазону физиологических особенностей [3]. Так, роговой слой эпидермиса сухой, кислый и имеет температуру ниже 37 градусов, что не в пользу бактериального роста, но эти условия выдерживает резидентская нормальная флора [18].
Но иногда Candida из комменсала трансформируется в агрессивный патоген, начиная экспрессировать так называемые факторы вирулентности. К этим факторам относятся морфология клетки, фенотипическое переключение, факторы адгезии и внеклеточная липолитическая и протеолитическая активность [11]. Известно, что факторы питания, такие как избыток или дефицит определенных питательных веществ, могут изменить эндогенную микробную флору [3]. Главная цель микроорганизма не состоит в том, чтобы вызвать смерть или даже повреждение хозяина, но выжить и репродуцировать, во время чего повреждение может быть только побочным эффектом [11].
У Candida существует специфический датчик аминокислот, который одновременно является их транспортером и назван «трансцептор», который приводит к морфогенезу и вирулентности. То, что именно азот является целью этого трансцептора, подтверждается тем, что экспрессия этого датчика подавляется в питательной среде, содержащей аммоний как хороший источник азота [3]. Отмечено также, что гены секретируемых протеиназ (как факторов вирулентности) индуцируются при отсутствии в среде экзогенного белка или пептидов [11], то есть в ответ на ограничение азота грибы начинают экспрессию доминантов вирулентности [3].
Вероятная функция секретируемых протеиназ – приобретение питания. Показано, что протеиназы были необходимы и достаточны, чтобы позволить быстрый рост Candida в питательных средах, содержащих белок как единственный источник азота [11]. Показано также, что внеклеточные матричные и поверхностные белки хозяина, такие как кератин, коллаген, ламинин, фибронектин, муцин, слюнной лактоферрин, альфа2-макроглобулин, почти все иммуноглобулины, комплемент и так далее, были эффективно гидролизированы секретируемыми протеиназами Candida [11, 12]. Необходимо отметить, что все секретируемые протеиназы Candida являются аспартат протеиназами, что внеклеточные ни серина, ни метало, ни цистеин протеиназы не были идентифицированы в патогенных штаммах Candida и что большинство аспартат протеиназ являются активными только в кислых условиях [11].
Таким образом, учитывая предположение, что свободные аминокислоты рогового слоя могут быть источником азота для Candida комменсала, можно предположить, что уменьшение содержания этих кислот или же их исчезновение могло бы быть инициирующим стимулом для индукции экспрессии факторов вирулентности и трансформации Candida в агрессивный патоген.
За продукцию свободных аминокислот в роговом слое эпидермиса ответственна каспаза14 – специфическая аспартат протеиназа, экспрессия которой ограничена главным образом ороговевающим эпителием. Каспаза14 обнаружена только у наземных млекопитающих, экспрессируется и активируется в эпидермисе, отсутствует в большинстве других тканей и важна для правильной деградации филаггрина на свободные аминокислоты и обслуживания эпидермальной гидратации. Отсутствие экспрессии каспазы14 в неороговевающем эпителии слизистой полости рта и в кератиноцитах ногтя подтверждает предназначение ее для генерации свободных аминокислот в роговом слое эпидермиса [8]. На основании принадлежности каспазы14 к аспартат протеиназам можно предположить определенную связь – при снижении активности аспартат протеиназы эпидермиса возникает активность аспартат протеиназы Candida .
Созревание каспазы14 наблюдается в эпидермисе путем протеолитического распада, в активацию ее вовлечена сериновая протеиназа и активация каспазы14 встречается во внутренней поверхности между зернистым и роговым слоями эпидермиса [8].
В эпидермисе идентифицировано целое семейство сериновых протеиназ – калликреинов (KLKs), из них вся хемотрипсин-подобная активность приписана KLK7, а трипсин-подобная активность на 50% обеспечивается KLK5, остальная идентифицирована как KLK14, причем KLK7 ответственна за главную часть полной протеолитической активности в роговом слое [5].
KLK5 – почти единственный фермент, найденный в эпидермисе, который может активировать KLK7. KLK5 мог активировать pro-KLK14. KLK14 мог активировать pro-KLK5, но не pro-KLK14. Активация KLK7 и KLK14 KLK5 происходила быстрее при кислом рН. Аутоактивация KLK5 и активация pro-KLK5 KLK14 оптимальна при рН 7-9 [5]. То есть в данном случае имеет место протеолитический каскад, в котором уже активированные ферменты служат активаторами других ферментов. Этот каскад может выглядеть следующим образом:
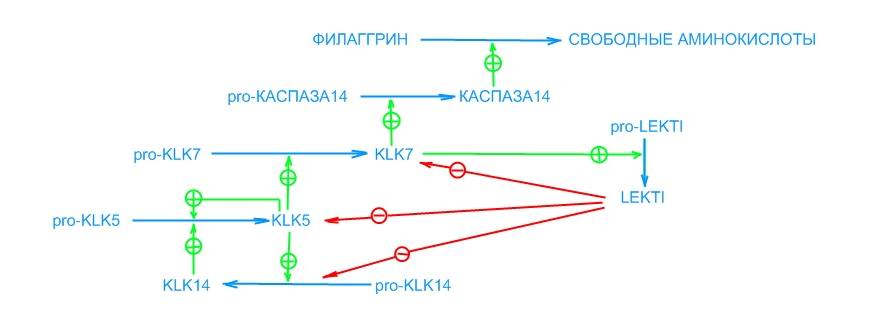
Весь этот каскад начинается во внеклеточном пространстве между зернистым и роговым слоями, когда рН меняется от нейтрального к кислому.
На уровень активности KLKs влияет несколько факторов. Так, 1,25(О)2VitD3, ретиноевая кислота и Са2+ индуцируют активность KLK5 и KLK7 в кератиноцитах [14]. Известно, что уровень относительной влажности влияет на активность каспазы14. Активность каспазы14 увеличивается, когда уровень влажности снижается менее 80% [21]. Это может быть связано с уровнем Са2+ - при низкой влажности уровни Са2+ увеличивались и, соответственно, увеличивалась активность KLKs, и наоборот (см. «Об ихтиозах…»). Этим, очевидно, можно объяснить проявление кандидоза в первую очередь в кожных складках: повышенная влажность вызывает снижение уровня Са2+, что влечет за собой снижение активности KLKs и, соответственно, каспазы14, что приводит к снижению содержания в роговом слое свободных аминокислот.
Во внеклеточное пространство между зернистыми и ороговевающими клетками поставляется мультидоменный лимфо-эпителиальный Казал-типа 1 ингибитор протеиназы LEKTI [13, 18]. Он со-локализуется с KLK7 в мембранных областях нормального рогового слоя и ингибирует KLK5 и KLK7 [18], а также KLK14 [4], таким образом влияя на десквамацию кератиноцитов. Со своей стороны KLK7 процессирует LEKTI, продуцируя активные одно и мультидоменные фрагменты LEKTI [4, 18], являясь, по сути, оптимизатором своей собственной продукции. Таким образом, в эпидермисе существуют сложные высокорегулируемые взаимосвязанные процессы десквамации и формирования водного барьера. Можно думать о том. что повышение активности LEKTI может повлечь за собой снижение уровня свободных аминокислот в роговом слое эпидермиса.
Показано, что физиологический статус хозяина является первичным фактором, контролирующим этиологию кандидоза. Небольшие изменения этого физиологического состояния могут трансформировать обычно безопасного дрожжевого сотрапезника в агрессивный патоген. Иммунокомпрометированные люди, такие как с ВИЧ-инфекцией, нейтропенией, пациенты интенсивной терапии и получающие иммуносупрессивные препараты после трансплантации органов, испытывают некоторую форму кандидозной инфекции слизистых оболочек [11]. К этому контингенту относятся и новорожденные [3].
В эпидермисе иммунная система представлена клетками Лангерганса. Они обычно расположены в парабазальном слое кератиноцитов, но их отростки достигают зернистого слоя. Считается, что клетки Лангерганса контролируют терминальное дифференцирование кератиноцитов.
Известно, что сразу после рождения начинается протеолиз филаггрина после перехода от водной среды в матке к сухой послеродовой среде [20]. Известно также, что в первую неделю после рождения в эпидермисе наблюдается взрыв пролиферации и дифференцирования клеток Лангерганса, что увеличивает их число 10-20-кратно и устанавливает сеть клеток Лангерганса. После первой недели жизни уровень их пролиферации снижается [7].
Показано, что ультрафиолетовое облучение уменьшает количество клеток Лангерганса в эпидермисе, инициируя их миграцию к подкожным лимфатическим узлам и способствуя иммунодепрессии [24]. Известно также, что ультрафиолетовое облучение вызывает утолщение рогового слоя эпидермиса и гиперпигментацию, которая обусловлена увеличением числа слоев кератиноцитов, тогда как количество пигмента на одну клетку остается прежним (см. «О мелатонине»). Можно думать о том, что увеличение количества слоев клеток может быть опосредовано снижением активности KLKs, что, в свою очередь, может быть вызвано увеличением активности LEKTI .
При атопическом дерматите наблюдается увеличение пролиферации и, соответственно, числа клеток Лангерганса в эпидермисе [7], а также отмечается снижение антипротеазной активности и, как результат, увеличение активности KLKs, что ускоряло деградацию корнеодесмосом (см. «Об ихтиозах…»).
Все перечисленное позволяет предположить, что наличие клеток Лангерганса в эпидермисе может быть связано с активностью LEKTI, то есть клетки Лангерганса могли бы супрессировать активность LEKTI, а их уменьшение в эпидермисе могло бы способствовать увеличению активности LEKTI и, соответственно, подавлению активности KLKs. В таком случае уменьшение числа клеток Лангерганса в эпидермисе может вызвать уменьшение уровня свободных аминокислот в верхних слоях рогового слоя и способствовать трансформации Candida в агрессивный патоген.
Кандидоз часто сопровождается гипопаратиреозом в комплексе аутоиммунных эндокринных и неэндокринных нарушений, называемом аутоиммунным полигландулярным синдромом тип 1 (APS-1) или аутоиммунная полиэндокринопатия-кандидоз-эктодермальная дистрофия (APECED) [6, 23]. Чаще всего представлены следующие состояния: кожно-слизистый кандидоз, гипопаратиреоз и болезнь Аддисона [6]. Кандидоз обычно начинается в течение первых двух лет жизни (до 5 лет), гипопаратиреоз проявляется в течение первого десятилетия жизни и является самой частой и иногда единственной эндокринной болезнью у пациентов APECED, надпочечная недостаточность присоединяется до возраста 15 лет (4-12), но иногда может проявиться в 20 лет [6, 16]. У женщин широко распространены нарушения овуляции [16].
Слизистый кандидоз, кроме стоматита, часто проявляется кандидамикозным кольпитом. Показано, что почти три четверти всех здоровых женщин испытывают, по крайней мере, одну влагалищную инфекцию дрожжей [11]. Известно, что влагалищный эпителий является многослойным, плоским, неороговевающим, но сквамозным эпителием. Известно также, что KLKs определяются во влагалищном стратифицированном сквамозном эпителии и уровни KLKs зависят от эстрогенов и достигают максимума после овуляции [22]. Очевидно, что снижение уровня эстрогенов могло бы способствовать развитию кандидоза.
Показано, что KLKs (в частности KLK13) сильно иммуноэкспрессированы паращитовидной железой [17]. Также обнаружено, что в паращитовидной железе экспрессируется и LEKTI [18]. Показано также, что в главных клетках паращитовидной железы процессинг предшественников паратгормона (РТН) происходит в протеосомах, в состав которых входят участки трипсин-подобной и хемотрипсин-подобной активности, что соответствует KLK5 и 7. Обработка клеток паращитовидной железы ингибиторами протеосом вызвала накопление предшественников РТН, задержку процессинга pro-РТН и уменьшила секрецию интактного РТН [19].
APECED связан с мутациями единственного гена, названного аутоиммунным регулятором (AIRE) [23]. AIRE является сильным активатором транскрипции и экспрессируется в медуллярных эпителиальных клетках тимуса, но не в тимоцитах, а также в дифференцированных дендритных клетках [16]. AIRE регулирует аутоиммунитет, вызывая эктопическую экспрессию периферическими тканями ограниченных антигенов в медуллярных эпителиальных клетках тимуса [23], но никакая значительная экспрессия AIRE не замечена в целевых органах аутоиммунной деструкции. AIRE зависит от наличия тимусных гематопоэтических клеток и от нормальной тимусной микроархитектуры. Экспрессия многочисленных ткань-специфических генов уменьшена или отменена у мышей AIRE-/- [16].
Показано, что в тельцах Гассаля тимуса присутствует каспаза14 [8], профилаггрин и филаггрин [9], KLKs (KLK13) [17] и LEKTI [18]. В эпителии слизистой ротовой области также присутствует профилаггрин и филаггрин [2]. Все перечисленные данные могут быть представлены в виде таблицы:
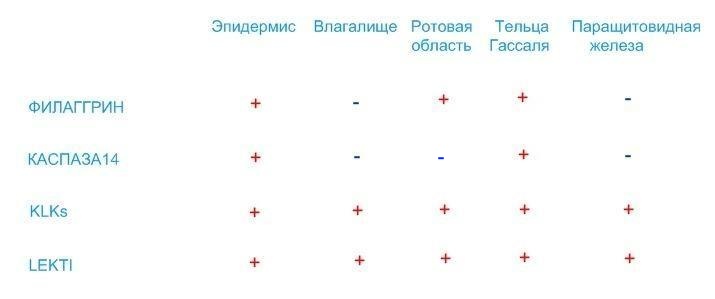
На основании таблицы можно предположить следующее: во-первых, присутствие KLKs и их регулятора LEKTI во всех представленных тканях может свидетельствовать о наличии в этих тканях целей для сериновых протеаз, в первых четырех это десквамация независимо от того, ороговевающий это или неогоровевающий эпителий (тельца Гассаля, как известно, формируются из десквамированных клеток медуллярного эпителия тимуса), а в паращитовидной железе – это процессинг РТН; во-вторых, наличие всех представленных соединений в эпидермисе и тельцах Гассаля может свидетельствовать о родстве этих тканей, а также о том, что при формировании телец Гассаля, помимо десквамации, может происходить и расщепление филаггрина на аминокислоты, что может иметь некоторую физиологическую уместность; и, в-третьих, отсутствие каспазы14 в эпителии слизистых оболочек полости рта и влагалища может свидетельствовать о том, что инициация кандидозного процесса в этих тканях может иметь отличия от эпидермиса, несмотря на участие в ней дифференцированных дендритных клеток.
Дело в том, что в эпидермисе сериновые протеазы KLK5 и KLK7 участвуют в ферментативном процессинге кателицидина предшественника белка hCAP18, продуцируя карбокси-терминальный пептидный фермент LL-37, способный убивать широкое разнообразие микроорганизмов [14]. Процессинг этими протеазами кателицидина не требует стимулов и приводит к конститутивному наличию LL-37 на поверхности кожи [1]. Видимо, этим можно объяснить повышенную предрасположенность кожи к инфекциям после интенсивного и пролонгированного ультрафиолетового облучения. Очевидно, что в слизистых влагалища и рта также продуцируются подобные бактерицидные ферменты, в этом процессе участвуют KLKs, а при миграции из слизистой дендритных клеток при иммунодефиците уровень этих защитных ферментов может снижаться.
Возникает ряд вопросов. Почему наблюдается такая последовательность – сначала кандидоз, далее гипопаратиреоз, а лишь затем болезнь Аддисона? Почему при кандидозе развивается еще и гипопаратиреоз, так как известно, что снижение Са2+ подавляет продукцию KLKs? Почему антитела к паращитовидной железе, присутствующие в сыворотке пациентов APS-1, обнаруживают также и при спорадических нозологических формах [16]? Почему ген AIRE экспрессируется лишь в медуллярных эпителиальных клетках тимуса и дендритных клетках и при нарушении этой экспрессии развивается APS-1 и в первую очередь кандидоз? Но это уже совсем другая тема.
Итак, на основании вышеприведенных фактов и умозаключений можно предположить, что кожно-слизистый кандидоз может быть следствием уменьшения присутствия дифференцированных дендритных клеток и, соответственно, увеличения антипротеазной активности, в эпидермисе или эпителии слизистых оболочек.
ЛИТЕРАТУРА:
1. Abtin A, Eckhart L, Mildner M, Ghannadan M, Harden J, Schroder J-M, Tschachler E. Journal of Investigative Dermatology 2009 129, 2193-2201
2. Berthelot JM, Maugars Y, Prost A, Youinou P. Rev Rhum Engl Ed. 1995 Feb;62(2):127-38
3. Biswar S, Van Dijck P, Datta A. Microbiol Mol Biol Rev 2007 June;71(2):348-376
4. Borgono CA, Michael IP, Komatsu N, Jayakumar A, Kapadia R, Clayman GL, Sotiropoulou G, Diamandis EP. The Journal of Biological Chemistry 2007 Feb 9, 282, 3640-3652
5. Brattsand M, Stefansson K, Lundh C, Haasum Y, Egebrud T. Journal of Investigative Dermatology 2005 124, 198-203
6. Buzi F, Badolato R, Mazza C, Giliani S, Notarangelo LD, Radetti G, Plebani A, Notarangelo LD. The Journal of Clinical Endocrinology and Metabolism 2003 Vol 88, No 7, 3146-3148
7. Chorro L, Sarde A, Li M, Woollard KJ, Chambon P, Malissen B, Kissenpfennig A, Barbaroux J-B, Groves R, Geissman F. JEM 2009 Dec 7, Vol 206, No 13 3089-3100
8. Denecker G, Ovaere P, Vandenabeele P, Declercq W. J Cell Biol 2008 Feb 11;180(3):451-458
9. Favre A. Acta Anat ( Basel ) 1989;135(1):71-6
10. Ghannoum MA. Clin Microbiol Rev 2000 Jan 13(1):122-143
11. Hube B, Naglik J. Microbiology 2001 147, 1997-2005
12. Karkowska-Kuletta J, Rapala-Kozik M, Kozik A. Acta Biochimica Polonica 2009, Vol 56, No 2, 211-224
13. Magert H-J, Standker L, Kreutzmann P, Zucht H-D, Reinecke M, Sommerhoff CP, Fritz H, Forssmann W-G. The Journal of Biologcal Chemistry 2007 Feb 9, 282, 3640-3652
14. Morizane S, Yamasaki K, Kabigting FD, Gallo RL. J Invest Dermatol 2010 May;130(5):1297-1306
15. Nailis H, Kucharikova S, Ricicova M, Van Dijck P, Deforce D, Nelis H, Coenye T. BMC Microbiol. 2010;10:114
16. Petersson P, Pitkanen J, Sillanpaa N, Krohn K. Clin Exp Immanol 2004 March; 135(3):348-357
17. Petraki CD, Karavana VN, Diamandis EP. J Histochem Citochem 2003 Apr 1, Vol 51 No 4, 493-501
18. Roelandt T, Thys B, Heughebaert C, De Vroede A, De Paepe K, Roseeuw D, Rombaut B, Hachem J-P. International Journal of Cosmetic Science 2009 August Vol 31, Issue 4, P 247-254
19. Sakwe AM, Engstrom A, Larsson M, Rask L. The Journal of Biological Chemistry 2002 May 17, 277, 17687-17695
20. Sandilands A, Sutherland C, Irvine AD, McLean WHI. Journal of Cell Science 2009, 122, 1285-1294
21. Schmuth M, Gruber R, Elias PM, Williams ML. Adv Dermatol 2007;23:231-256
22. Shaw JL, Petraki C, Watson C, Bocking A, Diamandis EP. Biol Chem 2008 Dec;389(12):1513-22
23. Trebusak Podkrajsek K, Milenkovic T, Odink RJ, Claasen-van der Grunten HL, Bratanic N, Hovnik T, Battelino T. European Journal of Endocrinology 2008 Vol 159, Issue 5, 633-639
24. Wang L, Jameson SC , Hogquist KA. The Journal of Immunology 2009 Nov 1, Vol 183, No 9, 5548-5553
В начало
На главную
© Дмитрий Марфунин