Дмитрий Марфунин
"О боковом амиотрофическом склерозе "
26 июня 2012 года
Как известно, боковой амиотрофический склероз (amyotrophic lateral sclerosis (ALS)) является фатальным, позднего начала, быстро прогрессирующим нейродегенеративным заболеванием, приводящим к мышечной слабости и атрофии, которые развиваются в паралич. Приблизительно 10% случаев являются семейным ALS (FALS), остальные 90% случаев – спорадические ALS (SALS) [12].
Невропатология ALS главным образом ограничена поражением моторных ядер ствола мозга и передних рогов спинного мозга [6]. Патология верхних моторных нейронов обозначена депопуляцией клеток Беца в двигательной области коры головного мозга, вариабельным астроцитарным глиозом и аксональной потерей пирамидального моторного пути с глиозом кортикоспинальных трактов. Патология нижних моторных нейронов прежде всего затрагивает моторные нейроны вентрального рога спинного мозга и ствола мозга. Количество нижних моторных нейронов может быть сокращено на 50%, остальные нейроны истощены и содержат внутринейронные включения [51]. Нейроны исчезают тихо, главным образом из-за апоптоза, и в клеточную смерть в ALS вовлечены ключевые элементы нормального апоптического пути [44, 51].
Отмечено также, что при ALS изменение дистальных моторных аксонов среди самых ранних патологических сдвигов, и процесс объяснен как “dying back”, то есть «отмирание» [1]. У пациентов нейротрубочки и нейрофиламенты в моторных нейронах истощены [44]. В большинстве аксональных включений моторных нейронов пациентов ALS найдены белки нейрофиламентов вместе с периферином [51]. При ALS увеличены серологические уровни нейрофиламентов, что является мерой аксональной дегенерации [28]. Разрушение цитоскелета медленного аксонального транспорта – одно из самых ранних патологических событий [1, 43]. Существует гипотеза, что клинические симптомы следуют из-за повреждения дистального моторного аксона, а не из-за активации пути клеточной смерти в клеточных телах [43].
Также показано, что при ALS нервно-мышечная денервация начиналась перед активацией апоптических белков [43]. А также что денервация терминальной пластинки отмечалась до признаков потери переднего корешка или тела клетки и до появления активированной микроглии, окружающей пораженные моторные нейроны [1].
Обращает на себя внимание то, что SALS начинается, поражая одну нейронную территорию, после ее повреждения вовлекая другую область рядом с первой; эта картина соблюдала соматотропную организацию области двигательной коры головного мозга. На территории нижних моторных нейронов прогрессия болезни, выраженная мышечной слабостью и атрофией, обычно линейно распространяется от одного метамера до другого в его окрестности [44]. 2/3 пациентов с типичным ALS имеют спинальную форму болезни (начало с конечностей) с фокальной слабостью и истощением мышц, где симптомы начинаются или дистально или проксимально в верхних и нижних конечностях [51].

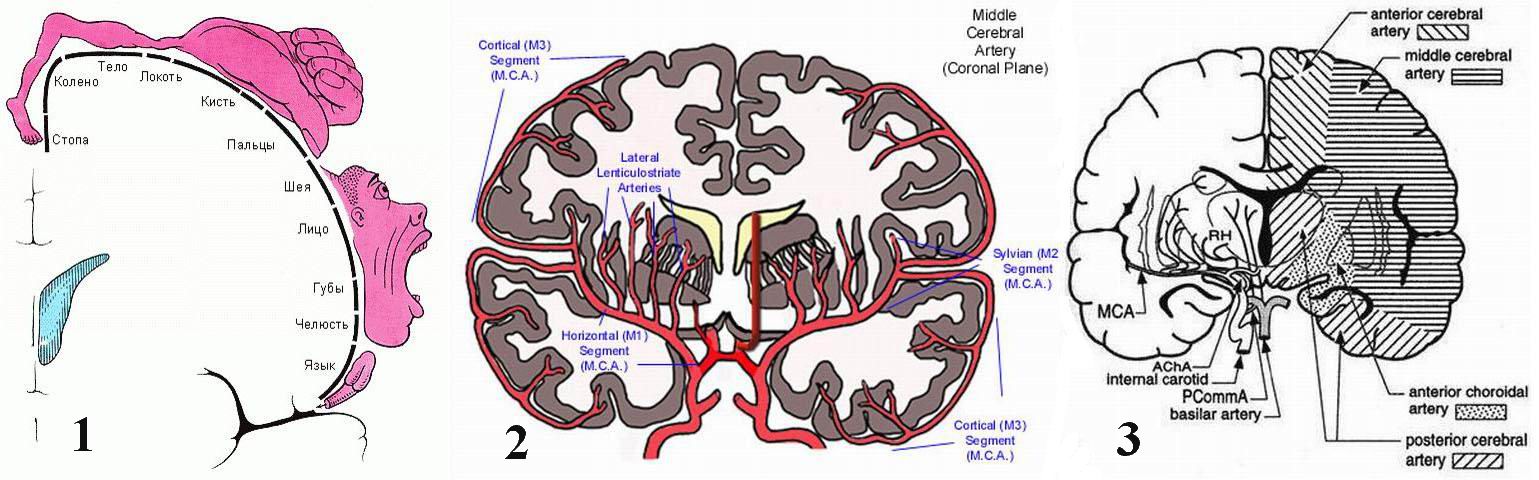
Как известно, верхние моторные нейроны расположены в двигательной области коры головного мозга – прецентральной извилине. Мышцы тела имеют определенную проекцию на этой извилине (см. рис 1). Известно также, что эта извилина получает артериальную кровь из двух артерий – передней мозговой и средней мозговой артерии (см рис 2). Граница зон кровоснабжения передней и средней артерии лежит приблизительно между проекцией тела и головы (см. рис 3). Соблазнительно предполагать, что поражающий агент (если такой существует) мог проникать в извилину извне. Учитывая вышеописанную картину прогрессии болезни, можно предполагать, что этот агент должен быть живым, способным к самовоспроизводству и мог бы лежать в основе и быть причиной болезни. Но были безуспешными попытки обнаружить вирусные частицы и молекулы в пределах моторных нейронов, корковых или спинальных, или в пределах тканей скелетных мышц. Более того, при рассмотрении пула моторных нейронов пациентов SALS в пределах пораженной области обнаруживается лишь недостаточность нейронов, но никакого намека на какую-либо борьбу нейрона против предполагаемого внедрившегося агента, способного его разрушить, только умеренная воспалительная реакция в окружающих тканях, главным образом основанная на наличии реактивной микроглии [44].
Если все же предположить, что такой агент существует, то возникает несколько вопросов. Во-первых, кто он? Во-вторых, почему поражает лишь моторные нейроны? Далее, почему поражает весь моторный тракт от терминальной пластинки до верхних моторных нейронов? Почему этот тракт поражается не одновременно, а начинается с терминальной пластинки и далее процесс распространяется вверх, создавая картину «отмирания»? И, наконец, почему моторные нейроны, не сопротивляясь, запускают процесс апоптоза?
Если, в качестве ответа на один из этих вопросов, предположить, забегая вперед, что моторный тракт поражается с целью (если такая существует) достижения гипокинезии, то для этого достаточно разрушить терминальную пластинку или, в крайнем случае, аксон переднего рога. Зачем же поражается весь тракт вплоть до клеток Беца?
Как известно, ALS ассоциируется с мутациями фермента CuZn-супероксид дисмутазы 1 (SOD1). Адекватно экспрессируемая SOD1 является главным цитоплазматическим антиоксидантом, нормальная функция которого – каталитическое конвертирование высокореактивной суперокиси (кислород с дополнительным электроном) или к перекиси водорода или к кислороду [15]. На экспрессии мутантной SOD1 основаны животные модели ALS. Но и в этих моделях отмечено, что высокий уровень экспрессии мутантной SOD1 в моторных нейронах не достаточен для раннего начала болезни [15]. Чтобы вызвать болезнь, мутантная SOD1 должна быть экспрессирована и в моторных нейронах и в окружающих клетках [30]. SOD1-null мыши не развивают ALS [13].
В человеческом же ALS мутации SOD1 составляют лишь малую пропорцию субъектов [28]. Мутации в SOD1 обнаружены лишь в 6% всех ALS [12]. В SALS мутации SOD1 встречаются только в 2% случаев [51].
Одними авторами обнаружены высокие уровни транскриптов SOD1 в спинном мозге, стволе мозга и лимфоцитах пациентов SALS [44]. Другими же не найдено различий в количестве SOD1 между пациентами ALS и контролем [12]. По активности SOD1 в эритроцитах различают 2 группы: большинство показало возрастное снижение функции, меньшинство показало увеличенную активность фермента [44]. Считается, что болезнь вызвана приобретенной токсичностью мутантных белков, а не уменьшенной активностью SOD1 [15], так как многие мутации SOD1, связанные с болезнью, не изменяют ее активности [13]. Более того, селективное вырезание активной мутантной SOD1 производит существенное ускорение поздней фазы болезни [15].
Показано, что у всех пациентов ALS в спинальных мотонейронах обнаруживаются меленькие зернистые SOD1-иммунореактивные включения [12]. Неправильно свернутая SOD1 была обнаружена во включениях спинного мозга субпопуляции пациентов SALS и аномальная свернутость и агрегация SOD1 могла бы быть общей патологической связью между SALS и FALS [10]. Включения SOD1 главным образом цитозольные, частичная со-локализация с катепсином D указывает, что включения становятся нацеленными для деградации через лизосомальный путь. Зернистые включения, содержащие неправильно свернутую SOD1, как правило, существуют в мотонейронах и SALS и FALS пациентов, испытывающих недостаток в мутантных SOD1 [12].
Оксидативный стресс – системное расстройство в SALS [44] и является одним из заметных обнаружений в ЦНС и периферической циркуляции пациентов ALS [1]. У пациентов SALS увеличен уровень окислительных маркеров в биологических жидкостях. Прогестерон, обеспечивающий антиоксидантную емкость, увеличен в сыворотке пациентов SALS [44] .
SOD1 дикого типа может приобрести свойства мутантных форм SOD1 через окислительное повреждение. Описано наличие оксидного дикого типа SOD1 в моторных нейронах спинного мозга SALS [1]. Существует гипотеза, что оксидантный стресс и окислительное повреждение SOD1 дикого типа могли быть позже начала SALS [13].
Несмотря на то, что оксидативный стресс – системное расстройство, и SOD1 – повсеместно экспрессируемый фермент, удивляет селективная уязвимость областей моторных нейронов к токсическим эффектам мутантных (и окисленных) форм адекватно широко экспрессируемой активной SOD1 [12, 15]. Можно предположить, что появление окисленных форм SOD1 и агрегация их в зернистые включения является следствием какого-то предшествующего события в системе моторного тракта.
Известно, что при SALS в области моторных нейронов наблюдается увеличение уровня концентрации глутамата (моторные нейроны, как известно, являются глутаматергическими нейронами). Увеличение глутамата не связано ни с интенсивностью нейронного повреждения, ни с распространением клинического компромисса, наблюдаемого в SALS [44]. Это связывают с уменьшением главного транспортера глутамата в мозге ЕААТ2, который селективен для астроглии и служит для очищения глутамата [44, 51]. Сверхстимуляция глутаматных рецепторов ведет к массивному притоку Са в нейроны [24, 44, 51]. Но наличие глутамата в высоких концентрациях в ЦНС не ограничена SALS, другие первичные дегенеративные нарушения ЦНС (Альцгеймера, Паркинсона, Хантингтона) также показывают высокие уровни глутамата в специфических областях, где нейроны стрессированы различными условиями [44].
Известно также, что при иммобилизации конечности (например, при переломе) в моторной области представления мышц данной конечности наблюдается перестройка – эти моторные нейроны испытывают гиперстимуляцию [53]. Можно предположить, что сверхстимуляция моторных нейронов в SALS имеет ту же природу. Если это так, то повышенная возбудимость развивается после наступления гипокинезии, то есть после денервации мышц.
Как известно, митохондрии являются очень динамичными органеллами и изобилуют в областях активных нейронов с интенсивными требованиями АТФ, таких как синаптические области. Они активно транспортируются и определяют субклеточные распределения, которые изменяются согласно физиологическим потребностям. При ALS моторные нейроны показали митохондриальные расстройства в иннервированной нейромышечной передаче в начале нервно-мышечной денервации. В моторных нейронах митохондрии играют важную роль в кратковременной обработке быстрого цитозольного Са++ перехода [43]. У пациентов ALS митохондриальные изменения, состоящие из набухания и увеличенного Са, присутствуют в терминалах моторного аксона на ранних стадиях [1, 43, 51]. Исследования показали дегенерированные митохондриальные вакуоли в аксонах и дендритах моторных нейронов в предсимптомных мышах. У пациентов SALS были найдены плотные конгломераты митохондрий в переднем роге поясничного спинного мозга и проксимальных аксонах. Считается, что митохондриальные расстройства могут быть вторичными эффектами, вызванными компрометированным аксональным транспортом. Эта гипотеза могла бы объяснить накопление дисфункциональных митохондрий в дистальных терминалах аксона и дегенерацию аксона, которые наблюдались в очень ранней стадии предсимптомных мышей ALS [43].
Дикого типа SOD1, также как и ее медный шаперон, была найдена в межмембранном пространстве митохондрии. Мутантная SOD1 также была найдена в межмембранном пространстве, матриксе и внешней мембране митохондрий. Вакуолизация митохондрий в мышах ALS была вызвана расширением межмембранного пространства и внешней мембраны, дегенерированные вакуоли были ограничены мутантной SOD1, которая со-локализовалась с митохондриальной внешней мембраны маркерами [43].
В связи с тем, что вакуолизация митохондрий и увеличение уровня Са встречается в одно и то же время, трудно определить, что в данном случае первично. Но при ALS обнаруживают также ингибирование генов кальциевых каналов [30] и снижение в нейронах уровней цитозольных кальций-связывающих белков, способных регулировать Са [44]. Возможно, что это может быть следствием сверхстимуляции глутаматных рецепторов [24, 51], но учитывая, что денервация терминальной пластинки отмечается до появления активированной микроглии, можно думать о том, что первичным в данном случае можно считать увеличение уровней Са в митохондриях. Увеличенный Са, как известно, может вызвать появление реактивных радикалов кислорода [43] и вызвать окисление дикого типа SOD1, которая начинает проявлять свойства мутантной SOD1 и может вызвать вакуолизацию митохондрий. Как известно, митохондрии самые критические структуры, которые при поражении оксидативным стрессом сами являются главным источником реактивных радикалов кислорода. Интересно, что изменения митохондрий под оксидативным стрессом при SALS не ограничены моторными нейронами, подобные изменения найдены в печени, мышце, лимфоцитах и коже [44]. То есть это вряд ли связано с глутаматной гиперстимуляцией.
Таким образом, можно думать о том, что увеличение уровня Са локально в аксонах и терминальных пластинках может вызвать оксидативный стресс, разрушение аксонального транспорта, дегенерацию митохондрий и, как результат, нервно-мышечную денервацию.
Отмечено, что у человека более 50% поясничных мотонейронов погибло спустя несколько лет после ампутации ноги [23] (картина похожа на состояние спинальных моторных нейронов при ALS (см. [51])). Показано также, что после ампутации руки моторные нейроны цервикального переднего рога были уменьшены в количестве не только на стороне ампутации, но и на противоположной стороне, но в меньшей степени. Ретроградная транснейрональная дегенерация наблюдалась также и в связующих нейронах после дегенерации клеток переднего рога, вызванной ампутацией [46].
Можно думать о том, что при нервно-мышечной дегенерации происходит, по сути, функциональная ампутация мышц. При этом нарушается обратная связь между моторными нейронами и их целевыми мышцами. Как и при фактической ампутации, очевидно, что это может привести сначала к гиперстимуляции мотонейронов, а затем к инициации процесса апоптоза.
Как известно, поставка глутамата в синапсах моторных нейронов побуждает изменения в пределах астроцитов, которые передаются их сосудистым ножкам, чтобы получать субстраты и приспособить кровоток согласно метаболическим требованиям нейронов [44]. Изобилие глутамата приводит к нейронной эксцитотоксичности из-за чрезмерного нейронного разряда и соответствующего увеличенного притока Са [24], то есть сверхстимуляция глутаматных рецепторов ведет к массивному притоку Са в нейроны [51].
При ALS нейроны не умирают одни: нейронное повреждение не клеточно-автономное и зависит от хорошо организованного диалога между моторными нейронами и микроглией. Изменений в пределах самого нейрона недостаточно, чтобы вызвать смерть моторного нейрона, но требует моторный нейрон-микроглия сигнализации по крайней мере на уровне сомы клетки [1]. Диалог между моторными нейронами и микроглией первоначально защищает моторные нейроны [1], также как и с астроглией [5]. Активация астроглии и микроглии встречается рано в патогенезе болезни [33, 44].
Но микроглиальная активация увеличивается в течение болезни [3]. Сигнализация от моторных нейронов вызывает микроглиальный выпуск реактивных радикалов кислорода и провоспалительных цитокинов, далее увеличивая стресс моторного нейрона и клеточное повреждение, и инициирует самораспространяющийся цикл повреждения моторного нейрона и клеточной смерти. Микроглия переключается от антивоспалительной и нейропротективной к провоспалительной и нейротоксичной [1].
Микроглию рассматривают как первичные иммунные клетки ЦНС [24]. Резидентная микроглия частично происходит из циркулирующих активированных моноцитов [25]. Активация микроглии заставляет их функционировать как антигенпредставляющие клетки и экспрессировать белки МНС класса II. Рекрутирование моноцитов и лимфоцитов вовлечено в патогенез ALS [5]. Активированные моноциты/макрофаги наблюдались у всех пациентов SALS, степень активности была непосредственно связана со скоростью прогрессии [54].
Показано, что Т клетки присутствуют на участках повреждения мотонейронов [5]. При ALS в областях деструкции моторного нейрона обнаружены инфильтрирующие иммунные клетки, включающие макрофаги, тучные клетки и Т клетки [28]. Показано, что Т клетки, В клетки, NK клетки, тучные клетки, макрофаги, дендритные клетки, микроглия, антигены, комплемент и цитокины участвуют в ограничении повреждения [8]. Хотя альтернативная Т клеточная инфильтрация может быть сопутствующим признаком, связанным с клиренсом погибших нейронов, возможно, что Т клетки могут индуцировать замедление деструкции моторного нейрона и облегчать регенерацию в ALS [14]. Поддерживается защитная роль специфической популяции Т клеток через контроль микроглиальной активации [24]. Т клетки могут влиять на моторные нейроны межклеточным контактом или секрецией цитокина или косвенно через активацию микроглии и макрофагов [14]. Наличие CD4+ T клеток морфологически и функционально изменяет микроглиальную и астроглиальную активацию, может поддержать нейропротекцию, модулируя глиальный баланс между трофикой и цитотоксичностью. При дефиците функциональных Т клеток у мышей ALS болезнь была ускорена, сопровождаясь неожиданно уменьшенными морфологическими маркерами глиоза, увеличенными уровнями мРНК для провоспалительных цитокинов и NOX2 и сниженными уровнями трофических факторов и глиальных глутаматных рецепторов. Донорского костного мозга предшественники Т клеток «обучаются» собственной локальной иммунной средой реципиента и, независимо от Т клеточного генотипа, организм направляет активацию этих трансплантированных лимфоцитов [5].
Исследования иммунной системы SALS показали значительные изменения в Т клетках периферической крови и циркулирующих моноцитах. В SALS увеличены уровни CD4+ T хелпера клеток и уменьшены уровни CD8+ T клеток; значительно уменьшены CD4+CD25+ регулирующие Т клетки и моноциты (CD14+) у больных в менее тяжелой стадии болезни; уменьшена экспрессия HLA-DR и моноцита хемоаттрактантного белка (МСР-1) рецептора (CCR2) на моноцитах как маркеров активации, что предполагает системную иммунную активацию [25, 28].
При ALS были увеличены компоненты комплемента С3, уровень IgG, уровни IL-13 и IL-17, уровни липополисахарида и С реактивного белка – все это свидетельствует о наличии иммунного ответа у субъектов ALS и предполагает системное воспаление [28]. Нейровоспаление в областях потери моторного нейрона очевидно в предсимптомных пациентах [29]. Но противовоспалительные лекарственные средства не замедляли прогрессию болезни [13]. Облучение тела, терапия стволовой клетки, внутривенные иммуноглобулины, циклофосфамид, имуран и преднизон не имели эффекта на течение болезни [28].
Роль иммунной сыворотки в ALS показана разрушенным барьером спинной мозг-кровь [10]. Микрокровотечения в пределах спинного мозга начинаются задолго до начала болезни в моделях ALS. Разрушение барьера спинной мозг-кровь сопровождается потерей плотных соединений между эндотелиальными клетками, что проявляется снижением уровней мРНК ZO-1 и occluding в спинном мозге пациентов ALS [15]. CD68 позитивные макрофаги могут проникать в спинной мозг с разрушенными ZO-1 соединениями [10]. CD4+ T хелперы клетки, которые поступали в спинной мозг на участках повреждения мотонейронов, нейропротективные [5].
Нарушением проницаемости гематоэнцефалического барьера можно было бы объяснить изменения концентрации цистатина С в цереброспинальной жидкости (ЦСЖ). Как известно, цистатин С – широко экспрессируемый ингибитор цистеиновой протеазы, уровень концентрации которого в норме в 5 раз выше в ЦСЖ, чем в плазме. Большинство цистатина С в ЦСЖ производится сплетением сосудистой оболочки [52]. Сплетение сосудистой оболочки при ALS не показывает воспалительных изменений [49]. Не обнаружена никакая мутация в гене цистатина С при ALS [50]. Уровни цистатина С в ALS были значительно увеличены в плазме и значительно уменьшены в ЦСЖ [27, 37, 52]. Уровни цистатина С непосредственно не коррелированны с дегенерацией моторного нейрона в ALS [52].
Цистатин С связан с ALS гистопатологически, поскольку он один из двух известных белков, локализующихся в тельцах Bunina [52]. Bunina тельца – интрацитоплазматические включения, положительно окрашивающиеся на цистатин и трансферрин и присутствуют в 70-100% случаев, представлены в клетках Беца и гипоталамических ядрах, редко замечены при других состояниях [51]. Цистатин С, ингибитор цистеиновой протеазы, вовлечен в белковую деградацию, является маркером телец Bunina в нижних моторных нейронах в ALS [31]. Уровни цистатина С в ЦСЖ изменяются во времени с увеличением при более медленной прогрессии [52]. Таким образом, можно предположить, что развитие болезни сопровождается блокадой ингибитора цистеиновой протеазы и, очевидно, связано с уровнем его активности, так как увеличение его уровня сопровождается замедлением прогрессии. Можно думать о том, что прогрессия болезни зависит от активности протеаз – чем меньше их активность, тем медленнее развивается болезнь, и наоборот. Это может быть подтверждено наблюдением, что при SALS происходит химическая ингибиция протеосом, в результате появляются накопления убиквитинированных и полуубиквитинированных белков, которые агрегируют в протеинные скопления, формирующие тельца включения [44]. Убиквитин-иммунореактивные включения почти универсальны в ALS, где могут быть замечены в 95% случаев, TDP-43 является главным белковым элементом [51]. TDP-43 – TAR (трансактивная реакция) ДНК связывающий белок, найден в ядре клетки, у него есть роль в трансляции ДНК в РНК [44].
Следствием увеличения уровня концентрации цистатина С в плазме можно считать изменения в коже у больных ALS. Как известно, пациенты ALS не развивают пролежни даже в терминальной стадии. Кожа на ощупь как загорелая кожа и теряет эластичность [36]. Эпидермис больных ALS иммунологически был положителен для цистатина С [35]. Известно, что в процессе десквамации клеток рогового слоя эпидермиса принимают участие протеазы. Можно думать о том, что увеличение концентрации цистатина С в плазме больных ALS может привести к блокаде этих протеаз и к ингибиции десквамации роговых клеток. Следствием чего может быть увеличение рогового слоя эпидермиса с вышеперечисленными свойствами.
Увеличение уровня концентрации цистатина С в плазме можно было бы объяснить разрушением барьера мозг-кровь. Но в уменьшении уровня концентрации цистатина С в ЦСЖ принимает участие не только разрушенный барьер мозг-кровь, но и блокада цистатина С в тельцах включения. Ингибицию протеазного ингибитора можно считать, по сути, предоставлением соответствующих условий для проявления активности протеаз. Но в то же время при SALS происходит ингибиция протеосом. Можно предположить, что благоприятные условия могли бы предоставляться каким-то иным протеазам.
Показано, что есть признаки депонирования в ЦНС при ALS иммуноглобулинов и комплемента. Иммуноглобулины от пациентов с ALS были токсичны к моторным нейронам в культуре. У мышей иммуноглобулины ALS вызывали расстройства в моторных концевых пластинках и вызывали дегенерацию моторных нейронов [28].
Показано также, что большинство пациентов с ALS обладают иммуноглобулинами, которые связываются с очищенными L-типа потенциал-зависимыми каналами кальция (VGCC). Кальция ионофора формирования альфа 1 субъединица VGCC идентифицирована как главный антиген VGCC, с которым связываются IgG ALS [19]. Правда, в других исследованиях не подтверждено присутствие антител против L-типа каналов кальция от скелетных мышц или мозга. Отмечается, что в ALS IgG препаратах присутствует увеличенная протеолитическая активность [2]. Протеазы могут быть частично ответственны за некоторые эффекты IgG ALS [34].
Можно думать, что в некоторых случаях антитела к кальциевым каналам все же могут определяться. Видимо, это может быть связано со стадиями болезни или с интенсивностью процесса. Возможно, что эти антитела продуцируются локально, быстро связываются с антигенами и могут не поступать в циркуляцию и не определяться в сыворотке. Во всяком случае, токсичность иммуноглобулинов к моторным нейронам вполне определенна.
Таким образом, учитывая все вышеизложенное, можно предположить, что иммунная система по какой-то причине воздействует на нейромышечную передачу, приводя к ее разрушению и к денервации мышц. Нарушение обратной связи между нейронами и мышцами вызывает гиперстимуляцию нейронов, активацию микроглии и, в конечном счете, запуск процесса апоптоза. Активация же микроглии приводит к повышению активности иммунной системы, направленной на нейропротекцию. Можно сказать, что иммунная система целенаправленно разрушает нейромышечное соединение, однако препятствует гибели моторных нейронов. Поражение нейромышечного соединения приводит к снижению двигательной активности, то есть к гипокинезии.
Известно, что у здоровых людей гипокинезия сопровождается острой гиперкортизолемией, значительным сокращением относительного и абсолютного числа общих лимфоцитов, CD3+ T клеток, субпопуляции Т хелперов и моноцитов, но увеличением CD8+ T клеток и NK клеток. Секреция IL-2 INF-гамма уменьшалась, а IL-4 была значительно более высокой [48]. При гипокинезии тимус и селезенка подвергается инволюции. В стадии адаптации гипокинетического стресса нормализации структуры и функции в лимфоидных органах не наблюдается. После гипокинезии во время периода реадаптации селезенка восстанавливается, но масса и структура тимуса не нормализуется [9].
Также гипокинезия увеличивала продукцию остеокласт-активирующего фактора кровяными мононуклеарными клетками, что коррелирует с большим количеством активных Т клеток [22]. Во время периода гипокинезии сывороточные концентрации Р, Са, Mg значительно увеличивались, тогда как концентрации интактного паратгормона и кальцитонина значительно уменьшались. Период постгипокинезии отмечался увеличением паратгормона и кальцитонина и уменьшением серологического Са [55].
Обращает на себя внимание некоторая парадоксальность реакции паратгормона и кальцитонина на гипокинезию. Если бы дело было лишь в повышении уровня Са в результате действия остеокласт-стимулирующего фактора, то увеличение Са должно было бы стимулировать кальцитонин и подавлять паратгормон, и наоборот. При гипокинезии же при повышении уровня Са уровни паратгормона и кальцитонина уменьшались. Очевидно, что на продукцию кальцитонина в данном случае мог бы влиять какой-то иной фактор. Так, например, известно, что на его продукцию отрицательно влияют мю-опиоидные пептиды.
При ALS же уровни сывороточного Са и паратгормона были повышены [18, 42]. Данных о сывороточном кальцитонине не обнаружено, но показано, что концентрация кальцитонина в ЦСЖ пациентов ALS была понижена [21]. Можно предположить, что паратгормон мог быть увеличен в результате увеличения уровня катехоламинов, которое наблюдается и в ЦСЖ и в крови у больных ALS [4].
Уровни кортизола пациентов ALS не отличались от контроля [40], лишь более раннее начало паралича коррелировало с более высокими уровнями кортизола [11]. Но у пациентов AKS наблюдается потеря циркадного ритма уровней кортизола [38]. Известно, что в норме уровень кортикостероидов ночью ниже, чем днем, утром возрастает, вечером снижается. Такая картина продукции кортикостероидов обусловлена секрецией мелатонина и опосредуется продукцией опиоидных пептидов (см. «О мелатонине», а также «О гомеостате»). Ночью мелатонин способствует повышению уровня опиоидов, которые ингибируют кортикотропин-релизинг гормон с соответствующим подавлением уровней кортикостероидов.
У пациентов ALS уровни кортизола в вечернем образце были значительно увеличены по сравнению с контролем [38]. Повышение уровней кортизола после пробуждения, то есть кортизола пробуждения реакция, была значительно меньшей у пациентов ALS [40]. Пациенты ALS также не показывали физиологического увеличения уровней кортизола после неожиданного умеренного стресса [38].
Так как в регуляции циркадного ритма кортикостероидов принимает участие опиоиды, можно думать о снижении активности опиоидергической системы у пациентов ALS .
Известно, что тиреотропин-релизинг гормон (TRH) ингибирует выпуск допамина и этим способствует увеличению выпуска пролактина. У пациентов ALS наблюдались нормальные оценки щитовидной железы [16, 17] и уровни пролактина были в пределах нормального диапазона [26]. После обработки пациентов ALS с TRH ответ пролактина был значительно уменьшен [20]. Как известно, выпуск допамина находится под тоническим ингибирующим влиянием опиоидных пептидов. Если при ALS активность опиоидергической системы снижена, то есть выпуск допамина не ингибирован, как в норме, то это могло бы объяснить меньший эффект введения TRH, чем в контроле. То есть это также можно расценивать как свидетельство снижения активности опиоидергической системы при ALS.
Показано, что при ALS у 2/3 пациентов наблюдается дефицит гормона роста [32, 39, 41]. Никакая корреляция не наблюдалась между пиковыми концентрациями гормона роста и возрастом, продолжительностью болезни, тяжестью или клинической формой [32]. Известно, что выпуск соматотропин-релизинг гормона находится под ингибирующим влиянием соматостатина, который, в свою очередь, находится под ингибирующим влиянием опиоидных пептидов. Иначе говоря, ингибиция ингибиции – это, по сути, стимуляция. То есть опиоидные пептиды способствуют выпуску соматотропин-релизинг гормона и, соответственно, гормона роста. Дефицит гормона роста при ALS также можно было бы расценить как снижение активности опиоидергической системы.
Как известно, средний возраст начала заболевания ALS между 50 и 60 годами [47]. Это возраст начала функционального угасания репродуктивной системы человека. Известно, что половые гормоны (в частности, эстрогены) положительно влияют на выпуск опиоидных пептидов. Можно думать о том, что при снижении уровня эстрогенов (а андрогены в мозге ароматизируются в эстрогены) может снижаться и активность опиоидергической системы.
Показано, что более высокий уровень дефицита гормона роста наблюдался у мужчин по сравнению с женщинами, с пиковой реакцией гормона роста у мужчин значительно ниже, чем у женщин [32]. Так же известно, что заболеваемость ALS у мужчин несколько выше, чем у женщин (отношение 1,5:1). Это также можно было бы объяснить более низкой активностью опиоидергической системы у мужчин-пациентов ALS.
Показано, что эндогенные опиоидные пептиды заняты в ауторегулирующих иммунных и нейроэндокринных процессах. Исследования демонстрируют, что эндогенные опиоидные пептиды показывают иммуностимулирующие действия [45]. Можно предположить, что снижение активности опиоидергической системы могло бы вызвать специфическую реакцию иммунной системы.
Показано также, что глутамин поддерживает оптимальную пролиферацию лимфоцитов и продукцию цитокинов лимфоцитами и макрофагами. Пониженная плазменная концентрация глутамина способствует иммуносупрессии [7].
Вышеперечисленные факты и умозаключения позволяют предположить, что конституционально сниженная активность опиоидергической системы, испытывающая дополнительно возрастное физиологическое снижение в результате угасания репродуктивной функции, могла бы вызвать специфическую реакцию иммунной системы, направленную на повышение уровня глутамина в крови.
Итак, боковой амиотрофический склероз можно было бы считать результатом заместительной активации глутаматергической системы иммунной системой в ответ на снижение активности опиоидергической системы.
ЛИТЕРАТУРА :
1. Appel SH, Zhao W, Beers DR, Henkel JS. Acta Myol. 2011 July;30(1):4-8
2. Arsac C, Raymond C, Martin-Moutot N, Dergent B, Couraud F, Pouget J, Seagar M. Ann Neurol. 1996 Nov;40(5):695-700
3. Barbeito AG, Mesci P, Boillee S. J Neural Transm 2010 Aug;117(8):981-1000
4. Barkhatova VP, Zavalishin IA, Kostiuk AV, Demina EG, Moskvitina TA. Zh Nevrol Psikhiatr Im S S Korsakova 1996;96(4):78-85
5. Beers DR , Henkel JS, Zhao W, Wang J, Appel SH. Proc Natl Acad Sci USA. 2008 Oct 7;105(40):15558-15563
6. Buttarelli FR, Circella A, Pellicano C, Pontieri FE. Eur J Neurol. 2006 Apr;13(4):416-8
7. Calder PC, Yaqoob P. Amino Acids. 1999;17(3):227-41
8. Calvo A, Moglia C, Balma M, Chio A. CNS Neurol Disord Drug Targets. 2010 Jul;9(3):325-30
9. Durnova GN, Kaplanskii AS. Arkh Anat Gistol Embriol. 1983 Aug;85(8):17-21
10. Fiala M, Chattopadhay M, La Cava A , Tse E, Liu G, Lourenco E, Eskin A, Liu PT, Magpantay L, Tse S, Mahanian M, Weitzman R, Tong J, Nguyen C, Cho T, Koo P, Sayre J, Martinez-Maza O, Rosenthal MJ, Wiedau-Pazos M. J Neuroinflammation. 2010;7:76
11. Fidler JA, Treleaven CM, Frakes A, Tamsett TJ, McCrate M, Cheng SH, Shihabuddin LS, Kaspar BK, Dodge JC. FASEB J. 2011 Dec;25(12):4369-77
12. Forsberg K, Jonson PA, Andersen PM, Bergemalm D, Graffmo KS, Hultdin M, Jacobsson J, Rosquist R, Marklund SL, Brannstrom T. PLoS One. 2010;5(7):e11552
13. Golden TR, Patel M. Antioxid Redox Signal. 2009 March;11(3):555-569
14. Holmoy T. Eur J Neurol 2008 Apr;15(4):360-6
15. Ilieva H, Polymenidou M, Cleveland DW. J Cell Biol 2009 Dec 14;187(6):761-772
16. Ilzecka J, Stelmasiak Z. Ann Univ Mariae Curie Sklodowska Med 2003; 58(1):343-7
17. Iwasaki Y, Kinoshita M. Jpn J Med 1989 May-Jun;28(3):309-11
18. Jackson CE, Amato AA, Bryan WW, Wolfe GI, Sakhaee K, Barohn RJ. Neurology 1998 Jun;50(6):1795-9
19. Kimura F, Smith RG, Delbono O, Nyormoi O, Schneider T, Nastainczyk W, Hofmann F, Stefani E, Appel SH. Ann Neurol. 1994 Feb;35(2):164-71
20. Klimek A, Szulc-Kuberska J, Stepien H. Neurol Neurochir Pol. 1990 Jan-Apr;24(1-2):31-6
21. Klimek A, Stepien H, Szulc-Kuberska J, Pisarek H. Pol Tyg Lek 1992 Apr 6-13;47(14-15):310-1
22. Konstantinova IV, Lesniak AT, Bozhikov NN, Uchakin PN. Kosm Biol Aviakosm Med. 1989 May-Jun;23(3):38-42
23. Kuno M. Rinsho Shinkeigaku. 1993 Dec;33(12):1275-7
24. Lasiene J, Yamanaka K. Neural Res Int. 2011;2011:718987
25. Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M, Bendotti C, Mora G. J Neuroimmunol 2009 May 29;210(1-2):73-9
26. Markianos M, Kosmidis ML, Sfagos C. Neuro Endocrinol Lett. 2006 Jun;27(3):355-8
27. Matias-Guiu J, Galan L, Garcia-Ramos R, Barcia JA, Guerrero A. Neurologia. 2010;25(6):364-373
28. McCombe PA, Henderson RD. Curr Mol Med. 2011 Apr;11(3):246-254
29. Moisse K, Strong MJ. Biochim Biophis Acta. 2006 Nov-Dec;1762(11-12):1083-93
30. Morahan JM, Yu B, Trent RJ, Pamphlett R. Amyotrophic Lateral Sclerosis 2009 Vol 10 No 5-6 Pag 418-429
31. Mori F, Tanji K, Miki Y, Wakabayashi K. J Neuropathpl Exp Neurol. 2009 Nov;68(11):1200-6
32. Morselli LL, Bongioanni P, Genovesi M, Licitra R, Rossi G, Martino E, Gasperi M. Clin Endocrinol (Oxf) 2006 Sep;65(3):385-8
33. Neusch C, Bahr M, Schneider-Gold C. Muscle Nerve 2007 Jun; 35(6):712-24
34. Nyormoi O. Ann Neurol. 1996 Nov;40(5):701-6
35. Ono S, Shimizu N, Imai T, Mihori A, Nagao K. Acta Neurol Scand. 2000 Jul;102(1):47-52
36. Ono S. Brain Nerve. 2007 Oct;59(10):1099-107
37. Pasinetti GM, Ungar LH, Lange DJ, Yemul S, Deng H, Yuan X, Brown RH, Cudkowicz ME, Newhall K, Peskind F, Marcus S, Ho L. Neurology 2006 Apr 25;66(8):1218-22
38. Patacchioli FR, Monnazzi P, Scontrini A, Tremante E, Caridi I, Brunetti E, Buttarelli FR, Pontieri FE. J Endocrinol Invest. 2003 Dec;26(12):RC23-5
39. Pellecchia MT, Pivonello R, Monsurro MR, Trojsi F, Longo K, Piccirillo G, Pivonello C, Rocco M, Di Somma C, Colao A, Tedeschi G, Barone P. Eur J Neurol. 2010 May;17(5);666-71
40. Roozendaal B, Kim S, Wolf OT, Kim MS, Sung KK, Lee S. Psychoneuroendocrinology 2012 Jan;37(1):20-6
41. Sacca F, Quarantelli M, Rinaldi C, Tucci T, Piro R, Perrotta G, Carotenuto B, Marsili A, Palma V, De Michele G, Brunetti A, Brescia Morra V, Filla A, Salvatore M. J Meurol. 2012 Jan;259(1):132-8
42. Sato Y, Honda Y, Asoh T, Kikuyama M, Oizumi K. Eur Neurol. 1997;37(4):225-9
43. Shi P, Gal J, Kwinter DM, Liu X, Zhu H. Biochim Biophis Acta 2010 Jan;1802(1):45-51
44. Sica REP, De Nicola AF, Gonzalez Deniselle MC, Rodriguez G, Gargiulo Monachelli GM, Martinez Peralta L, Bettini M. Arq Neuro-Psiquiatr 2011 Aug vol 69 no 4
45. Stefano GB, Kream RM. Arch Med Sci 2010 Jun 30;6(3):456-460
46. Suzuki H, Oyanadi K, Takahashi H, Ikuta F. Acta Neuropathol 1995;89(5):464-70
47. Ticozzp N, Tiloca C, Morelli C, Colombrita C, Poletti B, Doretti A, Maderna L, Messina S, Ratti A, Silani V. Archives Italiennes de Biologie 2011,149:65-82
48. Uchakin PN, Stowe RP, Paddon-Jones D, Tobin BW, Ferrando AA, Wolfe RR. Aviat Space Environ Med. 2007 Jun;78(6):608-12
49. Vercellino M, Votta B, Condello C, Piacentino C, Romagnolo A, Merola A, Capello E, Mancardi GL, Mutani R, Giordana MT, Cavalla P. J Neuroimmunol 2008 Aug 13;199(1-2):133-41
50. Watanabe M, Jackson M, Ikeda M, Mizushima K, Amari M, Takatama M, Hirai S, Ikeda Y, Shizuka-Ikeda M, Okamoto K. Brain Res. 2006 Feb 16;1073-1074
51. Wijesekera LS, Leigh PN. Orphanet J Rare Dis. 2009;4:3
52. Wilson ME, Boumaza I, Lacomis D, Bowser R. PLoS One 2010;5(12):e15133
53. Zanette G, Manganotti P, Fiaschi A, Tamburin S. Clin Neurophysiol. 2004 Jun;115(6):1264-75
54. Zhang R, Gascon R, Miller RG, Gelinas DF, Mass J, Hadlock K, Jin X, Reis J, Narvaez A, McGrath MS. J Neuroimmunol 2005 Feb;159(1-2):215-24
55. Zorbas YG, Kakurin VJ, Afonin VB, Charapakhin KP, Yarullin VL, Deogenov VA. Biol Trace Elem Res. 2000 Aug;76(2):113-131